宏基因组二、三代测序混合组装软件OPERA-MS
混合组装宏基因组实现高精度分析人体微生物组中的抗性基因和移动元件
Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes
Nature Biotechnology [IF:31.864]
2019-07-29 Articles
DOI: https://doi.org/10.1038/s41587-019-0191-2
第一作者:Denis Bertrand1
通讯作者:Niranjan Nagarajan1,7*
其它作者:Jim Shaw, Manesh Kalathiyappan, Amanda Hui Qi Ng, M. Senthil Kumar, Chenhao Li(李陈浩), Mirta Dvornicic, Janja Paliska Soldo, Jia Yu Koh, Chengxuan Tong, Oon Tek Ng, Timothy Barkham, Barnaby Young, Kalisvar Marimuthu, Kern Rei Chng, Mile Sikic
作者单位:
1 计算与系统生物学,新加坡基因组所(Computational & Systems Biology, Genome Institute of Singapore, Singapore, Singapore)
7 新加坡国立大学(National University of Singapore, Singapore, Singapore.)
热心肠日报
Nature子刊:宏基因组二、三代混合组装新软件OPERA-MS
创作:刘永鑫 审核:刘永鑫 08月02日
原标题:混合宏基因组组装实现人体微生物组中的抗性基因和移动元件的高精度分析
-
OPERA-MS采用重复感知聚类和精确的支架方法结合,实现二、三代序列的混合宏基因组组装;
-
基于模拟和真实宏基因组样本评估,获得目前最高质量的宏基因组,比长读长更高的碱基准确度,比短读长更高的连续性和比混合组装更少的错误,可获得低丰度物种的高质量基因组;
-
软件还可实现同一物种内菌株水平组装,获得稀有物种的高质量参考基因组;
-
结合纳米孔读长,实现80个完整质粒或噬菌体序列组装,为研究肠道抗生素抗性组精细研究提供可能。
二代测序通量高、准确度高,但读长短;三代测序读长长,但错误率高、成本高。将这两者的优势结合,目前在宏基因组领域还没有得到广泛应用,存在很多技术难题没有解决。近日,来自新加坡基因组所的Niranjan Nagarajan课题组发布了一款二、三代测序混合组装软件OPERA-MS,组装结果不仅碱基准确率高,而且短读长数据拼接长度提升了一个数量级。
OPERA-MS整合了宏基因组聚类和精确支架算法,基于虚拟肠道微生物组和人工群落数据测序,研究者仅用9×长读长覆盖深度组装出了接近目前最完整的宏基因组,也组装出低丰度(<1%)物种的高质量基因组。值得一提的是,OPERA-MS还可在亚种水平上获得基因组结果。将Nanopore测序应用于抗生素治疗病人的肠道宏基因组研究,发现长读长组装质量较短读长提升了200倍。这一重镑成果于7月29日发表于世界顶级期刊《Nature Biotechnology》。
摘要
通过高通量宏基因组测序已经实现了微生物组的组成分析。然而,现有方法不是设计用于组装来自短读长和长读长混合序列。我们提出了一个名为OPERA-MS的混合宏基因组组装软件,它将组装宏基因组采用重复感知聚类和精确的支架方法结合,实现精确地组装复杂群落。使用预定义的体外和虚拟肠道微生物组进行评估,OPERA-MS组装的宏基因组具有比长读长(> 5×; Canu)更高的碱基对准确度,比短读长更高的连续性(~10× NGA50; MEGAHIT,IDBA-UD) ,metaSPAdes)和比非宏基因组混合组装软件(2×; hybridSPAdes)更少的组装错误。OPERA-MS在同一物种的多个基因组存在下提供菌株分辨率的组装结果,可在~9倍长读取覆盖率下获得稀有物种的高质量参考基因组(<1%)。我们使用OPERA-MS组装28个抗生素治疗患者的肠道宏基因组,并显示包含长纳米孔读长产生更多连续组装(比短读长组装提高200倍),包括超过80个成环质粒或噬菌体序列和一个新的263 kbp巨型噬菌体。高质量的混合组软件可以对人类患者的肠道抗生素抗性组进行精细的观察。
Characterization of microbiomes has been enabled by high-throughput metagenomic sequencing. However, existing methods are not designed to combine reads from short- and long-read technologies. We present a hybrid metagenomic assembler named OPERA-MS that integrates assembly-based metagenome clustering with repeat-aware, exact scaffolding to accurately assemble complex communities. Evaluation using defined in vitro and virtual gut microbiomes revealed that OPERA-MS assembles metagenomes with greater base pair accuracy than long-read (>5×; Canu), higher contiguity than short-read (~10× NGA50; MEGAHIT, IDBA-UD, metaSPAdes) and fewer assembly errors than non-metagenomic hybrid assemblers (2×; hybridSPAdes). OPERA-MS provides strain-resolved assembly in the presence of multiple genomes of the same species, high-quality reference genomes for rare species (<1%) with ~9× long-read coverage and near-complete genomes with higher coverage. We used OPERA-MS to assemble 28 gut metagenomes of antibiotic-treated patients, and showed that the inclusion of long nanopore reads produces more contiguous assemblies (200× improvement over short-read assemblies), including more than 80 closed plasmid or phage sequences and a new 263 kbp jumbo phage. High-quality hybrid assemblies enable an exquisitely detailed view of the gut resistome in human patients.
主要结果
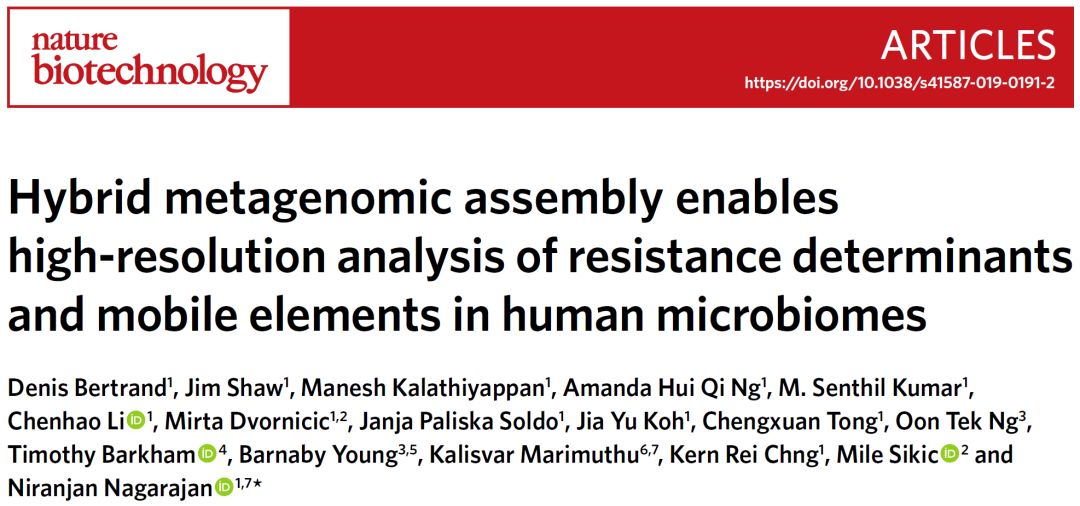
图1. OPERA-MS工作流程图
Fig. 1: OPERA-MS workflow.
首先将宏基因组的短读长拼接为重叠群,并将短读取和长读长比对至重叠群以获得覆盖信息和跨越序列(步骤1)。然后绑定跨越读长获得组装图中重叠群之间的边,该组装图表示整个宏基因组的连续性信息(步骤2)。将重叠群组织成层次聚类,其中重叠群之间的距离随基因组距离及其覆盖差异而增加(步骤3)。然后基于BIC(贝叶斯信息准则)将树切割成最佳簇(步骤4)。可选步骤,为了改善可获得参考基因组物种的聚类,计算每个聚类与完整细菌基因组数据库之间的Mash基因组距离(步骤5)。然后,如果在装配图中存在支持信息以形成物种特定的超级簇,则合并簇(步骤6)。进一步分析这些超级簇以解卷积来自可区分的亚种基因组的重叠群(步骤7)。最后,使用针对分离基因组的程序(OPERA-LG;步骤8),独立地构建每个簇并填充间隙。
Short reads are first assembled by a metagenomic assembler into contigs, and short and long reads are mapped to them to obtain coverage information and spanning reads (Step 1). Spanning reads are then bundled to get edges between contigs for an assembly graph that represents the contiguity information of the whole metagenome (Step 2). Contigs are organized into a hierarchical clustering where the distance between contigs increases with genomic distance and their difference in coverage (Step 3). The tree is then cut into optimal clusters based on the BIC (Step 4). Optionally, to improve the clustering for species where a reference genome is available, the Mash genomic distance between each cluster and a database of complete bacterial genomes is computed (Step 5). Clusters are then merged if there is supporting information in the assembly graph to form species-specific super-clusters (Step 6). These super-clusters are further analyzed to deconvolute contigs that come from distinguishable subspecies genomes (Step 7). Finally, each cluster is independently scaffolded and gap-filled using a program meant for isolate genomes (OPERA-LG; Step 8).
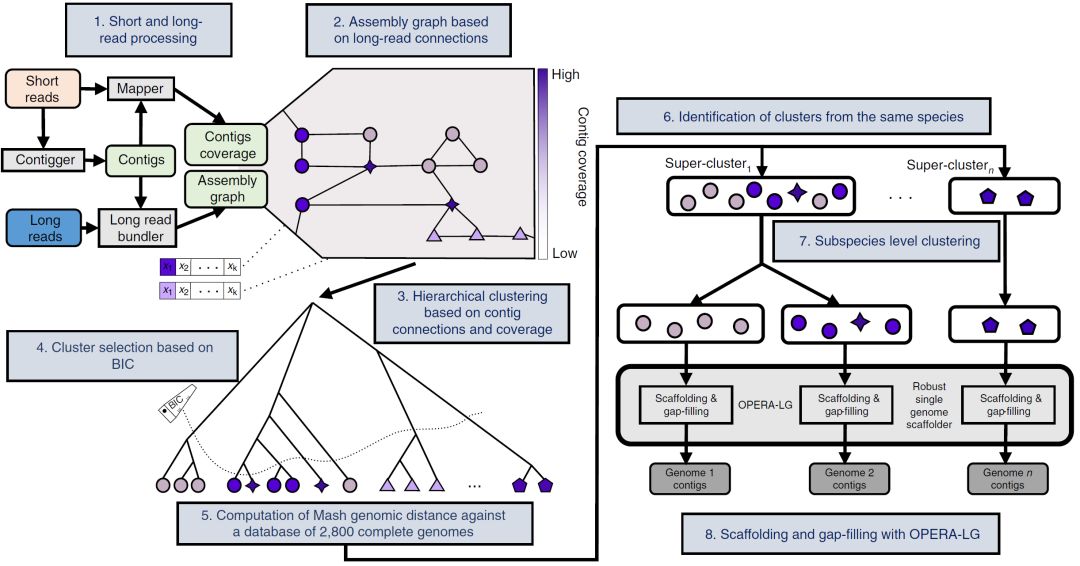
图2. 宏基因组数据混合组装基因组评测
Fig. 2: Benchmarking hybrid assembly of genomes from metagenomes.
a-c,作为短读长代表性组装软件metaSPAdes(a),长读长组装软件Canu(b)和混合组装软件OPERA-MS(c)的测序覆盖率增加与组装连续性的增加。请注意,混合装配在跨越覆盖方面有效改进了短读长和长读长的装配结果,可在低至9×长读长覆盖度下产生接近完整的基因组(NGA50 > 1 Mbp)。未组装的基因组显示为带有黑色边框的圆圈。d,OPERA-MS与其他组装软件相比较提高的装配连续性(NGA50)。对于MEGAHIT和IDBA-UD,组装基因组中覆盖度上升的数量为3,12,20和19,对于metaSPAdes和hybridSPAdes为3,13,21和19,对于Canu为4和16。请注意,Canu不会组装低覆盖率的基因组,因此在这些范围内不提供指标。数据以箱形图表示(中心线,中位数;箱限,上下四分位数; 须线,1.5×四分位数间距; 点,异常值)。e,不同组装软件的组装错误率,实线表示中值。除了hybridSPAdes之外,大多数组装软件每 Mbp(虚线)产生大约1个错误的组装。在每个部分中,每个数据点代表来自模拟群落的一个基因组。
a–c, Increase in assembly contiguity as a function of read coverage for a representative short-read assembler (a), long-read assembler (b) and hybrid assembler (c). Note that hybrid assembly improves over short- and long-read assembly in terms of scaling across coverage ranges and producing near-complete genomes (NGA50 >1 Mbp) with as little as 9× long-read coverage. Unassembled genomes are shown as circles with black borders. d, Improvements in assembly contiguity (NGA50) provided by OPERA-MS in comparison with other assemblers as a function of long-read coverage. The number of assembled genomes, in ascending order of coverage is 3, 12, 20 and 19 for MEGAHIT and IDBA-UD, 3, 13, 21 and 19 for metaSPAdes and hybridSPAdes and 4 and 16 for Canu. Note that Canu does not assemble low-coverage genomes and hence metrics are not provided in those ranges. Data are presented as box plots (center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, outliers). e, Misassembly rates for different assemblers, with solid lines indicating median values. Most assemblers produce ~1 large misassembly per Mbp (dashed line), except for hybridSPAdes. In each part, each data point represents one genome from the mock communities.
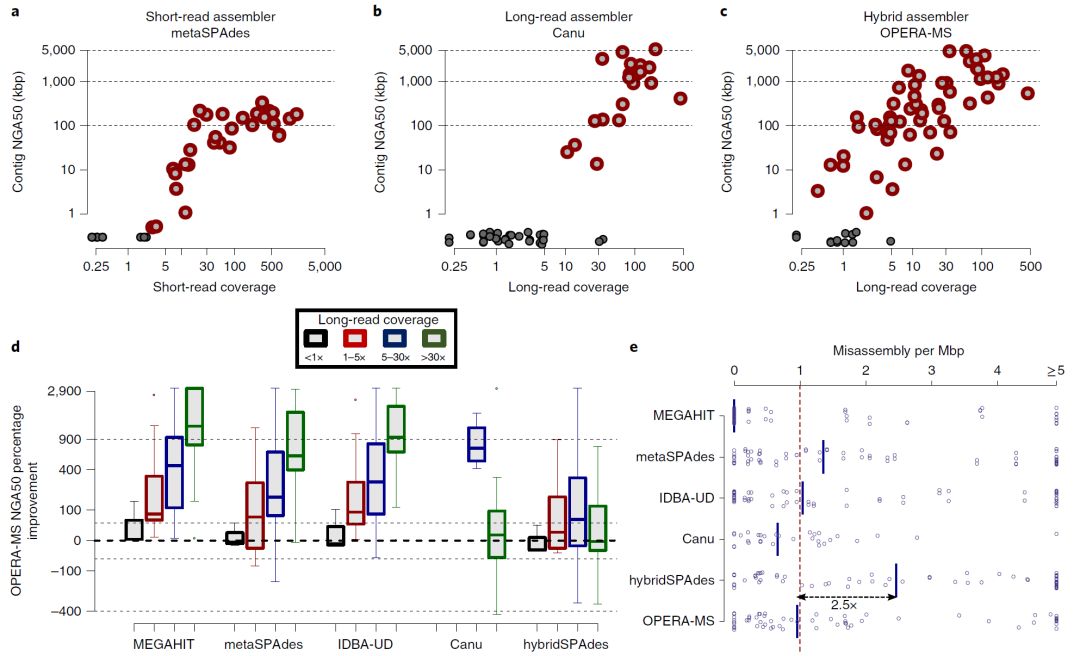
图3. 组装虚拟肠道微生物组
Fig. 3: Assembly of a virtual gut microbiome.
a,构建虚拟肠道微生物组,代表复杂的宏基因组数据集,同时保留评估组装与金标准参考的能力。
b,与不同覆盖范围内的其他组装软件相比,使用OPERA-MS获得组装连续性(NGA50)的改进情况。点代表在宏基因组中具有至少两个菌株的物种(在GIS20和S2中存在的物种,如MetaPhlAn2报道的丰度 > 0.1%(参考文献49)(v.2.6.0))。按照覆盖度的上升,组装的基因组的数量对于Canu是1,对于其他方法是2,6,4和5个。数据以箱形图表示(中心线,中位数;箱限,上下四分位数; 须线,1.5×四分位数间距; 点,异常值)。
c,不同组装软件的组装错误率(每个基因组一个点)的比较,实线表示中值。
d,在分箱后评估仅Illumina数据(M,MEGAHIT)和混合(H,hybridSPAdes; O,OPERA-MS)组装宏基因组组装以用于下游分析。包含最大部分参考基因组的区域(GIS20参考文献;具有粗体名称的物种在宏基因组中具有至少两个菌株)评估以下参数:(1)基因组完整性,在分箱中基因组的比例,(2)基因组纯度,分箱中碱基对应正确参考的百分比,(3)基因完整性,在分箱中完全组装的基因比例和(4)通路完整性,其组成基因超过90%的通路出现在组装的分箱中。
a, Construction of a virtual gut microbiome that represents a complex metagenomic data set while retaining the ability to evaluate assemblies against gold-standard references. b, Improvement in assembly contiguity (NGA50) obtained using OPERA-MS compared with other assemblers over different coverage ranges. Dots represent species that have at least two strains in the metagenome (species present in GIS20 and S2 with an abundance >0.1% as reported by MetaPhlAn2 (ref. 49) (v.2.6.0)). The number of assembled genomes, in ascending order of coverage, was 1 for Canu and 2, 6, 4 and 5 for the other methods. Data are presented as box plots (center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, outliers). c, Comparison of misassembly rates (one dot per genome) for different assemblers, with solid lines indicating median values. d, Evaluation of Illumina-only (M, MEGAHIT) and hybrid (H, hybridSPAdes; O, OPERA-MS) metagenomic assemblies after binning for their utility in downstream analysis. Bins that contained the largest fraction of a reference genome (GIS20 references; species with bold names have at least two strains in the metagenome) were evaluated for (1) genome completeness, the fraction of the genome represented in the bin, (2) genome purity, percentage of bases in the bin that correspond to the correct reference, (3) gene completeness, fraction of genes that were fully assembled in the bin and (4) pathway completeness, fraction of pathways with over 90% of their constituent genes being assembled and binned together.
图4. 移动元件和与人肠道微生物组中宿主物种的关联
Fig. 4: Mobile elements and association with host species in the human gut microbiome.
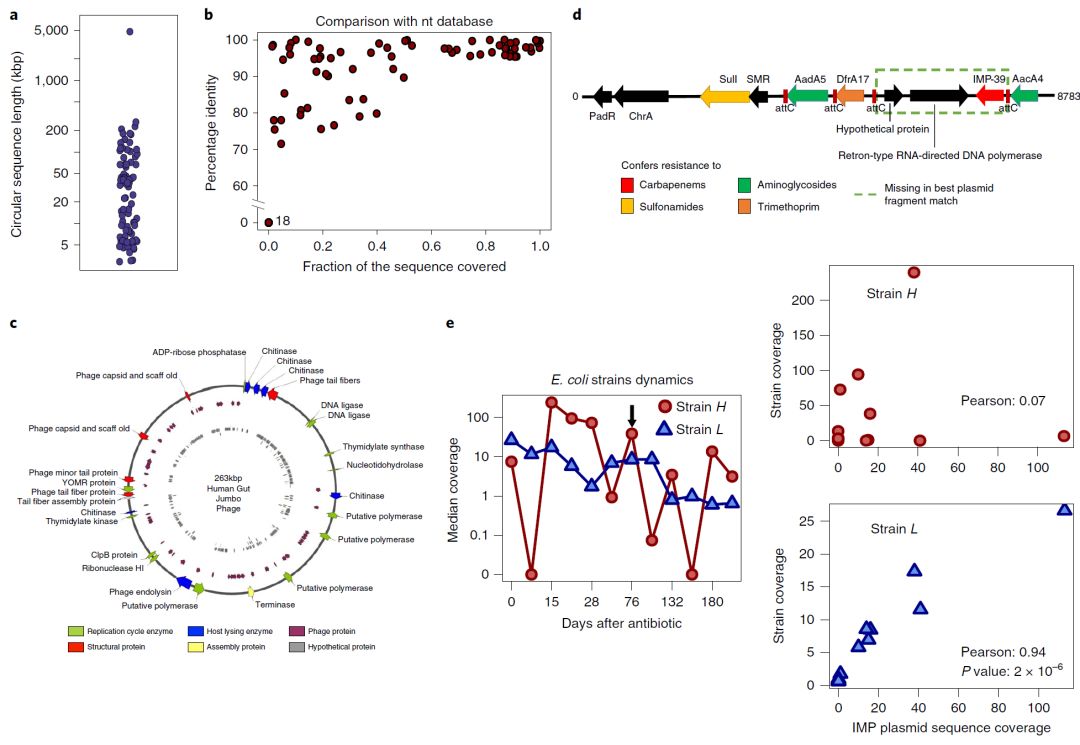
a,来自OPERA-MS的28个人肠道宏基因组数据集中完全组装成环序列的基因组大小分布,说明了组装不同大小和复杂性的环状基因组的能力(质粒,噬菌体和细菌基因组)。
b,与NCBI核苷酸(nt)数据库中的序列(基于BLAST搜索)比对,覆盖序列的比例与组装的环状序列的平均序列相似度。许多组装序列从端到端(右上角)显示出与已知序列的良好比对和相似度,但是一些仅具有局部相似性(左上角),并且一些似乎是新的(左下角; 18个序列) 。
c,观察到最大的新(在nt数据库中没有匹配)环状序列(263kbp)的注释,发现与噬菌体生命周期相关的蛋白,包括复制、组装和宿主裂解相关,表明组装的序列是假定的巨型噬菌体。
d,OPERA-MS从耐受碳青霉烯的肠杆菌科细菌定植患者的肠道微生物组中组装出新的多重抗性区域。除临床相关的碳青霉烯酶基因区域外,该区域还含有赋予氨基糖苷类、甲氧苄氨嘧啶和磺胺类抗性的基因,限制了治疗选择。
e,OPERA-MS菌株水平组装可以进行质粒与基因组基于跨越时间点的测序覆盖信息进行关联(n = 12)。左图:来自第76天的数据的杂合宏基因组装配中观察到的两种大肠杆菌菌株基因组的覆盖度的变化(黑色箭头)。右图:质粒覆盖度与两种大肠杆菌菌株之间的相关性表明它是可能含有IMP基因的质粒的菌株L使用R中的学生t-检验(双侧)计算P值。
a, Distribution of genomes sizes for fully assembled circular sequences from OPERA-MS in 28 human gut metagenome data sets, illustrating the ability to assemble circular genomes of varying sizes and complexity (plasmids, phages and bacterial genomes). b, Fraction of sequence covered versus average sequence identity of the assembled circular sequences in comparison to sequences in the NCBI nucleotide (nt) database (based on BLAST searches). Many of the assembled sequences showed good alignment and homology to known sequences from end to end (top right corner), but some only had local similarities (top left corner), and a few appear to be new (bottom left corner; 18 sequences). c, Annotation of the largest (263 kbp) observed new circular sequence (no matches in nt database) revealed proteins associated with a phage life cycle, including replication, assembly and host lysis, indicating that the assembled sequence is a putative jumbo phage. d, A new multiple resistance region assembled by OPERA-MS from the gut microbiome of a patient colonized by carbapenem-resistant Enterobacteriaceae. Apart from the clinically relevant carbapenemase gene cassette, the region also harbors genes that confer resistance to aminoglycosides, trimethoprim and sulfonamides, limiting treatment options. e, Strain level assembly with OPERA-MS enabled association of plasmid to genome based on correlation in read coverage across timepoints (n = 12). Left panel: Variation in coverage of two Escherichia coli strain genomes seen in the hybrid metagenomic assembly of data from day 76 (black arrow). Right panel: Correlation between the coverage of the plasmid and the two E. coli strains reveals that it is strain L that likely harbors the IMP gene containing plasmid. The P value was computed using Student’s t-test in R (two-sided).
总结
本文介绍了一种基于混合数据的宏基因组组装软件OPERA-MS,比较分析了其与其他几种短读长、长读长数据组装软件对宏基因组研究的效能。它能够显著的提升组装的连续性,并且还能够解决亚种级基因组的组装,解决了长读长数据的原始错误率、覆盖度问题和短读长数据的读长缺陷,即使对于低深度覆盖的数据也能有出色的表现。为了验证软件的应用能力,研究者还模拟了人体肠道微生物组的数据,发现其对于临床宏基因组、抗生素耐药性基因的研究上面也能提供较好的帮助。
Reference
-
Denis Bertrand, Jim Shaw, Manesh Kalathiyappan, Amanda Hui Qi Ng, M. Senthil Kumar, Chenhao Li, Mirta Dvornicic, Janja Paliska Soldo, Jia Yu Koh, Chengxuan Tong, Oon Tek Ng, Timothy Barkham, Barnaby Young, Kalisvar Marimuthu, Kern Rei Chng, Mile Sikic, and Niranjan Nagarajan. (2019). Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nature Biotechnology.10.1038/s41587-019-0191-2
-
有了OPERA-MS,人体肠道微生物不用愁!
相关阅读
-
NBT:宏基因组”读云”10X建库+雅典娜算法组装获得微生物高质量基因组
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读