单原子催化剂因其最大的原子使用率和电子状态的高度可调性,成为能源材料研究领域的热点,例如过渡金属单原子催化剂在电催化CO2还原上表现出优异特性,同时,单原子催化剂也被认为是在原子水平上揭示催化机制的理想模型系统。然而,单原子催化剂的实际应用受到稳定性差和转换效率低的限制,比如,用于CO2还原(CO2RR)的氮掺杂石墨烯上的镍单原子为CO生成提供了高催化活性和法拉第效率,但由于形成 *COOH 中间体的高能量势垒,其开启电位较大;铁单原子对CO2还原的开启电位较低,但铁位点与反应中间体(如*CO)的强结合严重降低了法拉第效率;过渡金属位点与给电子中间体的结合强度太强,同样会降低析氧反应的催化活性等等。
与单原子相比,异类双原子位点催化剂是利用两个相邻的不同金属原子来实现它们的功能互补和协同作用,特别是,与中间体的结合能可以通过两个相邻的异类金属之间的电子相互作用来调整,克服了单原子催化的不足。然而,合理地构建具有高催化性能和多功能性的双原子位点催化剂并揭示其工作机制仍然是一个巨大的挑战。
近日,中山大学材料科学与工程学院曾志平副教授研究组构建了在氮掺杂石墨烯上的高密度镍铁双原子高性能电催化体系,并对催化增强机制进行了深入的研究。他们通过光电子光谱,包括XAS、UPS和XPS,鉴定了两个异类金属原子的电子结构特征。X射线吸收近边结构和扩展X射线吸收精细结构测量证实了镍铁双单原子的镍电子态具有与单原子相似镍氮配位键,但其中铁电子态不同于铁单原子,呈现更高的K-edge电子结构,即更高的氧化态;小波变换扩展X射线吸收图谱显示了金属-氮和金属-金属键的存在,进一步证实了独特的双原子结构。理论计算进一步探究和证实了双金属原子对之间的d-d轨道耦合对二氧化碳还原和析氧的影响。镍和铁之间的d轨道(尤其是dz2, dx2-y2, 和dxz)相互作用,导致轨道能级下降和电子分布的离散性,从而有益于*CO中间体的脱附。异类双原子体系中,配位氮pz轨道在费米能级附近的电子态密度增加,得益于轨道耦合中铁电子转移至氮的pz轨道,使得铁活性中心的价态增加,从而有利于CO2RR和析氧反应。异类双原子催化剂结合了铁单原子(较低的开启电位)和镍单原子(较高的选择性催化)两者的优势,表现出远优于对应的单原子催化剂的卓越电催化活性、高度的选择性和稳定性。配备该催化剂的金属-CO2电池可在大电流密度下稳定和持续地充放电,在CO2还原的同时获取电能和燃料(CO,CH4, H2等),具有很好的实用性前景,有望拓展至火星(大气CO2浓度95%)探索。
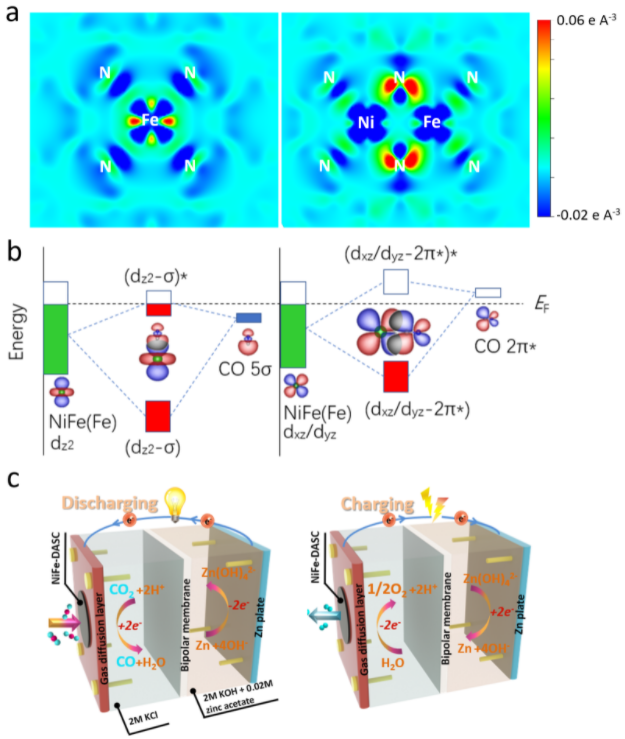
图1. Fe单原子(Fe-SAC)和FeNi 双原子位点(FeNi-DASC)催化剂中(a)Fe位点电荷分布,(b)Fe位点3d 轨道于吸附的CO (5σ and 2π* 轨道相互作用示意图,(c)FeNi-DASC的金属-CO2电池充放电过程。
该研究成果论文“Orbital coupling of hetero-diatomic nickel-iron site for bifunctional electrocatalysis of CO2 reduction and oxygen evolution”发表在国际重要学术期刊Nature Communications,曾志平副教授是论文的第一作者,新加坡南洋理工大学陈鹏教授、刘彬教授、苏州科技大学杨鸿斌教授为文章的共同通讯作者。该研究工作得到了国家自然科学基金委员会、中山大学“百人计划”启动经费和新加坡科技局、新加坡教育部的支持。
来源:中山大学
论文链接:
https://www.nature.com/articles/s41467-021-24052-5