为了实现精准医疗,许多国家已经开展本地人群的全基因组测序(WGS)研究。不过,目前的WGS资源主要来自欧洲人,而亚洲基因组在公共数据库中的代表性不足。
为此,新加坡科技研究局(A*STAR)领导的团队对新加坡的中国人、马来人和印度人三个群体开展了大规模测序,并在《Cell》杂志上发表了题为“Large-Scale
Whole-Genome Sequencing of Three Diverse Asian Populations in Singapore”的文章。
“新加坡三个主要种族提供了东南亚地区遗传多样性的独特快照,”作者在文中写道。这篇文章的通讯作者包括华中科技大学的王超龙教授和新加坡国立大学的刘建军教授,他们同时隶属于新加坡科技研究局。
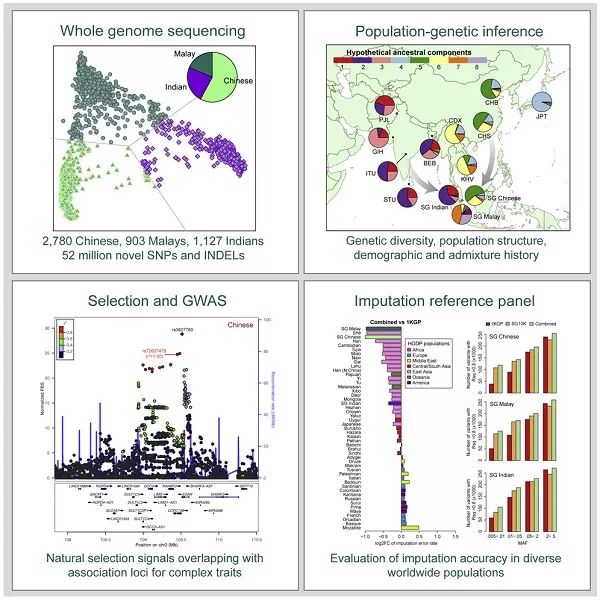
SG10K联盟的成员对新加坡的4,810名中国人、马来人和印度人进行了测序,发现了9,830万个新颖的或已知的多态性,可用来评估亚洲血统在这些族裔中的比例。他们还追踪了至少20个表现出自然选择迹象的基因座,其中14个与复杂性状和疾病相关联。
基于这些研究,作者认为“对新加坡人的全基因组测序有望让整个亚洲以及世界其他地区的人群受益”。他们预计,“SG10K的数据将成为宝贵的资源,可推动亚洲人的遗传性状和复杂疾病研究,并缓解目前人类遗传学研究中的群体差异”。
这项研究属于新加坡万人基因组测序计划的早期阶段。在此,研究人员对4,800多人进行测序,平均测度深度达13.7倍。参与者包括2,780名中国人、903名马来人和1,127名印度人,来自8个不同的队列。
在剔除了不符合质量控制标准的变异后,他们获得了8,910万个SNP和910万个小的插入缺失(indel)。其中,大约4,560万个SNP和630万个indel未曾出现在以往的群体研究中。
研究人员将1,200多名个体以往的基因型数据与此次分析的结果进行比较,确认了这些变异的质量,然后才深入研究芯片未覆盖的罕见变异。通过这种方式,他们记录了2,525名健康个体的变异数量和类型,包括与隐性疾病风险相关的变异。
通过主成分分析(PCA),研究人员发现新加坡中国人和新加坡印度人分别与东亚人和南亚人重叠,而新加坡马来人与其他群体相对较远,这意味着马来人代表了东南亚的土著人。同时,他们还追溯了新加坡各个人群的起源。
考虑到SG10K的数据将成为亚洲人群数据的宝贵资源,研究人员接着评估了数据集的有效性。“即使采用严格的标准,我们也能够检测到亚洲人群以往报道的许多基因座,”他们写道。这些结果凸显了SG10K数据将成为推动亚洲人群遗传学研究的宝贵资源。
原文检索
Large-Scale Whole-Genome Sequencing of Three Diverse Asian Populations in
Singapore
Cell VOLUME 179, ISSUE 3, P736-749.E15, OCTOBER 17, 2019
